Puesta al día
Antiagregantes plaquetarios y cirugía oral: retirar o no retirar, esa es la cuestión
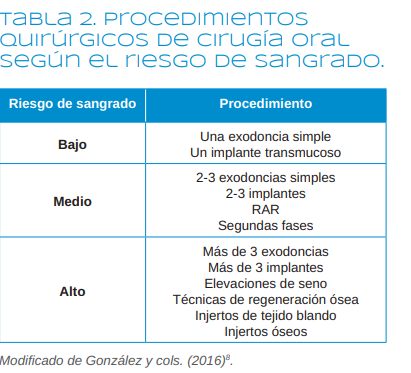
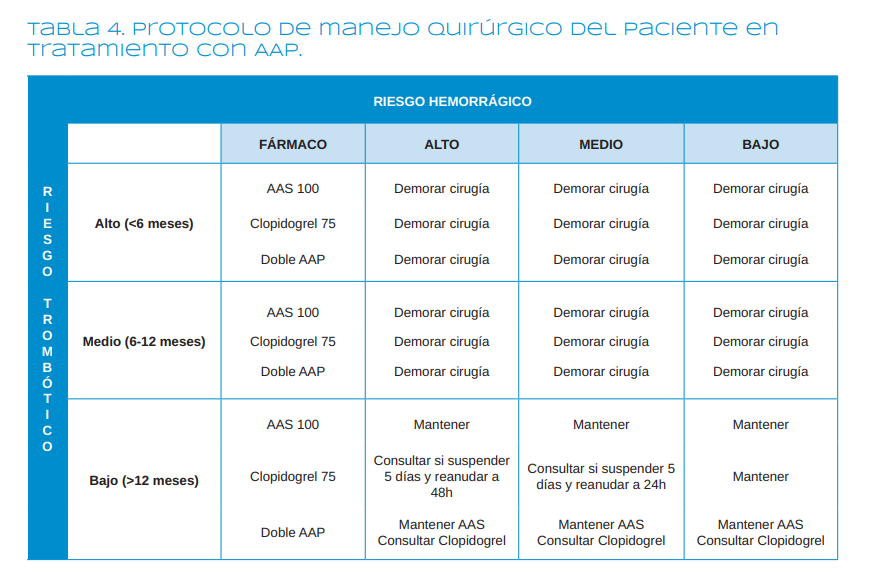
Las enfermedades cardiovasculares constituyen una de las patologías sistémicas más prevalentes en el mundo occidental. Muchos pacientes cardiópatas han tenido un episodio coronario agudo y están siendo tratados con antiagregantes plaquetarios. La terapia con estos fármacos puede suponer un reto para el odontólogo, que debe enfrentarse a un importante dilema: o mantener el fármaco, con el consiguiente riesgo hemorrágico, o retirarlo, con la posibilidad de que se produzcan complicaciones tromboembólicas, suponiendo un riesgo para la vida del paciente. Por ello, los odontólogos deberíamos conocer cuál debe ser el manejo de este tipo de pacientes ante la perspectiva de realizar un procedimiento quirúrgico en la cavidad oral o incluso una simple extracción dentaria.
Los objetivos de esta revisión narrativa son, en primer lugar, recordar la fisiología plaquetaria y los mecanismos de formación del trombo plaquetario; en segundo lugar, profundizar en los mecanismos de acción de los diferentes fármacos antiagregantes plaquetarios; y, en tercer lugar, ya que no existen guías clínicas al res- pecto, realizar un abordaje crítico de las pautas existentes para el manejo odontológico de este tipo de pacientes, en aras de prevenir la aparición de posibles complicaciones, no solo locales, sino, lo que es más importante, complicaciones sistémicas. En estos casos, antes de retirar la terapia antiagregante, convendría sopesar el riesgo hemorrágico versus el riesgo de generar un nuevo episodio tromboembólico, como puede ser la trombosis del stent o la recidiva del accidente coronario agudo, eventos que podrían poner en riesgo la vida del paciente.
Cardiovascular disease is one of the most prevalent systemic pathologies worldwide; those patients usually have had an acute coronary event which is treated with antiplatelet therapy. These drugs represent a challenge for the dentist, who must face a major dilemma: either maintain the drug, with the consequent bleeding risk, or withdraw it, with the possibility of thromboembolic complications, entailing a risk to the patient’s life. Therefore, dentists should know how to manage patients treated with these drugs when performing a surgical procedure or even a simple tooth extraction.
The objectives of this narrative review are, firstly, to recall platelet physiology and the mechanisms of platelet thrombus formation; secondly, to go more deeply into the mechanisms of action of the different antiplatelet drugs; and thirdly, since there are no clinical guidelines on this topic, to critically review the existing guidelines related to the dental management, in order to prevent the appearance of possible complications, not only local, but more importantly, systemic complications. In these cases, before interrupting antiplatelet therapy, the risk of bleeding should be evaluated against the risk of generating a new thromboembolic episode, such as stent thrombosis or recurrence of the acute coronary accident, events that could put the patient’s life at risk.
La enfermedad cardiovascular es la patología sistémica que genera mayor morbimortalidad en el mundo occidental. En España, según datos del Instituto Nacional de Estadística, en el año 2020, la patología cardiovascular fue la primera causa de muerte entre la población general, por delante de los tumores y de la COVID-19. En este sentido, la cardiopatía isquémica supone la principal causa de muerte en hombres y los accidentes cerebrovasculares en mujeres.
Dentro de las enfermedades cardiovasculares, la patología aterosclerótica es la principal causa de morbimortalidad en nuestro país, donde se incluyen los síndromes coronarios agudos, las enfermedades cerebrovasculares y la enfermedad arterial periférica1. El mecanismo etiopatogénico que subyace en todas estas patologías es la rotura de la placa de ateroma, que desencadena el fenómeno de la agregación plaquetaria, que es la causa del proceso trombótico agudo. Por ello, durante los últimos años, se han desarrollado nuevos fármacos antiagregantes plaquetarios, que constituyen la piedra angular de la prevención de la recurrencia de episodios isquémicos agudos, tanto a corto como a largo plazo. Estos fármacos merecen ser conocidos por el odontólogo, sobre todo a la hora de llevar a cabo un procedimiento quirúrgico, para saber cuál debe ser el correcto manejo odontológico para evitar posibles complicaciones, no solo desde el punto de vista del sangrado local, sino, lo que es más importante, para evitar complicaciones sistémicas (como trombosis del stent o aparición de un nuevo evento tromboembólico), que podrían poner en riesgo la vida del paciente 2
El propósito de esta revisión narrativa es, en primer lugar, recordar la fisiología plaquetaria y los mecanismos de formación del trombo plaquetario; en segundo lugar, profundizar en los mecanismos de acción de los diferentes fármacos antiagregantes plaquetarios; y, en tercer lugar, ya que no existen guías clínicas al respecto, realizar un abordaje crítico de las pautas existentes para el correcto manejo odontológico de los mismos, de cara a la realización de un procedimiento quirúrgico en la cavidad oral o incluso una simple extracción dentaria, con suficientes garantías de éxito.
Fisiología de la plaqueta
La plaqueta es uno de los elementos formes de la sangre, junto con los glóbulos rojos y los glóbulos blancos. Normal- mente, en la sangre hay entre 150.000 y 400.000 plaquetas por mL, y el volumen plaquetario medio suele ser de 7-9 micrómetros cúbicos. Las plaquetas proceden de las células madre hematopoyéticas de la médula ósea, concretamente de la estirpe mieloide y tienen una vida media de 7 a 10 días. En su interior, se encuentran los gránulos alfa y gránulos densos, donde se acumulan moléculas de especial relevancia en la fisiología plaquetaria 3
Las plaquetas juegan un papel primordial en la hemostasia, ya que inician la reparación de las lesiones vasculares, formando el tapón plaquetario, pero también promueven la coagulación de la sangre, a través de la activación de la trombina liberada desde las propias plaquetas y del calcio liberado de los gránulos densos que es necesario para la formación de la fibrina 3.
En la fisiología plaquetaria intervienen varias enzimas, como la ciclooxigenasa (COX), que transforma el ácido araquidónico (AA) procedente de los fosfolípidos de la membrana, en prostaglandinas (PG), que son el tromboxa- no A2 (TXA2), vasoconstrictor y proagregante plaquetario y la prostaciclina (PGI2), que es vasodilatadora y antiagre- gante y se origina en el endotelio vascular3. Otras enzimas de las plaquetas son: la fosfolipasa A2, que libera el AA de los fosfolípidos de la membrana y la fosfodiesterasa, que hidroliza el AMPc.
Las plaquetas tienen en su membrana receptores que son glicoproteínas (GP) y están inactivos en condiciones nor- males. Los más relevantes son GP Ia y GP VI que se unen al colágeno, GP Ib que se une al factor von Willebrand (FVW) y GP IIb/IIIa, que se une a varias proteínas, pero la más importante es el fibrinógeno. Estos receptores parti- cipan en los fenómenos plaquetarios de adhesión (unión de la plaqueta al vaso lesionado), activación (cambio de morfología de la plaqueta que provoca la secreción de los gránulos) y agregación (unión entre varias plaquetas)3.
Mecanismo de formación del trombo plaquetario
Las plaquetas circulan en el torrente sanguíneo en forma inactiva. Pero cuando se produce una lesión en un vaso, se expone el colágeno subendotelial, que es el estímulo para reclutar las plaquetas que van a formar el tapón pla- quetario. Hay que recordar que las plaquetas no se ad- hieren al endotelio intacto, pero sí se pueden adherir a un cuerpo extraño dentro del torrente sanguíneo (como puede ser, por ejemplo, un stent coronario o una prótesis valvular cardíaca)3.
Adhesión: Cuando se lesiona un vaso, las plaquetas circulantes enlentecen su velocidad sobre la zona dañada, en contra del flujo sanguíneo que las empuja, gracias a que el receptor plaquetario GP Ib se une al factor von Willebrand (FVW) de la matriz bajo el endotelio. Después, el colágeno subendotelial establece una unión más estable uniéndose al GP Ia y GP VI plaquetarios3.
Activación: Tras la adhesión, se produce la activación de la plaqueta, apareciendo en el exterior los receptores que estaban inactivos. Estos activan moléculas intracelulares, que provocan un cambio en la morfología plaquetaria, con emisión de pseudópodos y liberación de ciertas sustancias que promueven la agregación plaquetaria, perpetuando el proceso. Estas moléculas, conocidas como agonistas plaquetarios son: el TXA2, el ADP y la trombina. De todos ellos, el ADP es el más potente para reclutar plaquetas y propagar el trombo arterial, por lo que se le considera un amplificador de la activación plaquetaria.
Las plaquetas presentan en su superficie tres receptores para el ADP: P2Y1, P2Y12 y P2X. Cada uno induce distintas vías de señalización plaquetarias, pero el P2Y12 es el más importante, ya que favorece la liberación del contenido de los gránulos, el aumento del calcio intracelular, la generación del TXA2 y la activación del receptor GP IIb-IIIa, que es clave en la agregación plaquetaria. En consecuencia, el bloqueo del receptor P2Y12 plaquetario es crucial para inhibir la activación y la agregación plaquetarias e impedir, por tanto, la formación del trombo plaquetario. Por todo ello, durante los últimos años se han desarrollado nuevos fármacos ca- paces de bloquear este receptor1.
Liberación: Tras la activación, se produce la liberación de las moléculas almacenadas en los gránulos de las plaquetas. Las plaquetas activadas pueden liberar hasta 300 pro- teínas diferentes. De los gránulos alfa, se liberan proteí- nas homólogas a las del plasma (fibrinógeno, fibronectina, factor XIII, FVW) y proteínas específicas de las plaquetas (Factor plaquetario 4-FP4, tromboglobulina, P-selectina, PDGF (factor de crecimiento derivado de las plaquetas) y trombospondina). De los gránulos densos se libera ADP, ATP, calcio y serotonina (5-hidroxitriptamina o 5HT)3.
Agregación: Una vez atrapadas las plaquetas en la zona dañada, se produce el reclutamiento de nuevas plaquetas desde la circulación sanguínea, lo que se conoce como agregación plaquetaria. La activación del receptor GP IIb/ IIIa es la vía final que conduce a la agregación plaqueta- ria. Una vez activado, se une a sus ligandos, que tienen la secuencia de aminoácidos RGD (Arg-Gly-Asp o argini- na-glicina-aspártico), como el fibrinógeno, pero también el FVW, la fibronectina y la vitronectina. Este receptor es específico de las plaquetas y se une de forma bivalente al fibrinógeno, formando puentes de unión entre dos plaquetas3.
Respecto a los mecanismos que regulan la agregación
plaquetaria, están los favorecedores y los inhibidores. Así, favorecen la agregación plaquetaria el ADP, la trombina, el
colágeno, la adrenalina y el TXA2. Mientras que el AMPc,
GMPc y la PGI2 la inhiben, así como el óxido nítrico (NO), que además de ser antiagregante plaquetario, es considerado el mayor vasodilatador del organismo3
Fármacos antiagregantes plaquetarios (AAP)
Los antiagregantes plaquetarios (AAP) son fármacos que
inhiben la agregación plaquetaria, actuando como antitrombóticos. Suelen actuar de forma irreversible y no se puede monitorizar su función, pero se suele considerar que su efecto dura lo que dura la vida media de la plaqueta, es decir, entre 7 y 10 días1.
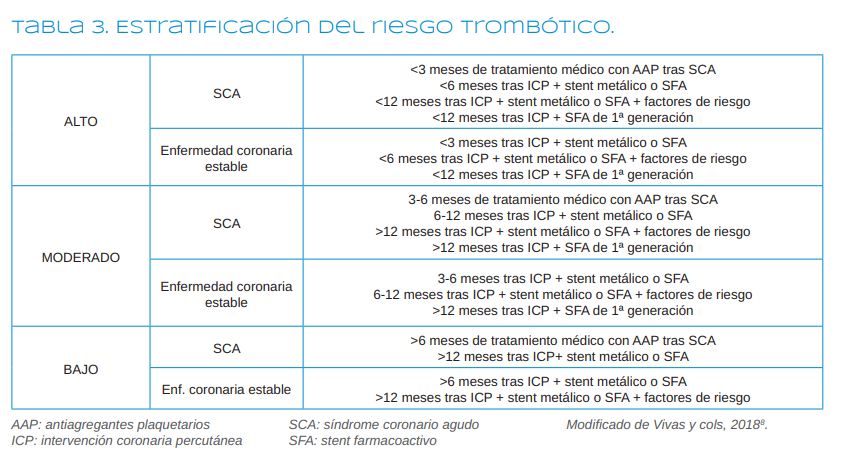
Las principales indicaciones de estos fármacos incluyen los síndromes coronarios agudos
(SCA) (que engloban el infarto agudo de miocardio-IAM y
la angina inestable), la enfermedad coronaria estable, la
enfermedad cerebrovascular y la enfermedad arterial periférica, pero también se utilizan después de los tratamientos quirúrgicos que se realizan tras la aparición de estos cuadros, como la intervención coronaria percutánea (ICP) o la cirugía de revascularización, y en la prevención de la recurrencia de los mismos, es decir, en la profilaxis secundaria de la enfermedad aterosclerótica 1 .
Los AAP son fármacos cuyo mecanismo de acción se basa
en inhibir las enzimas plaquetarias, como la COX (ácido
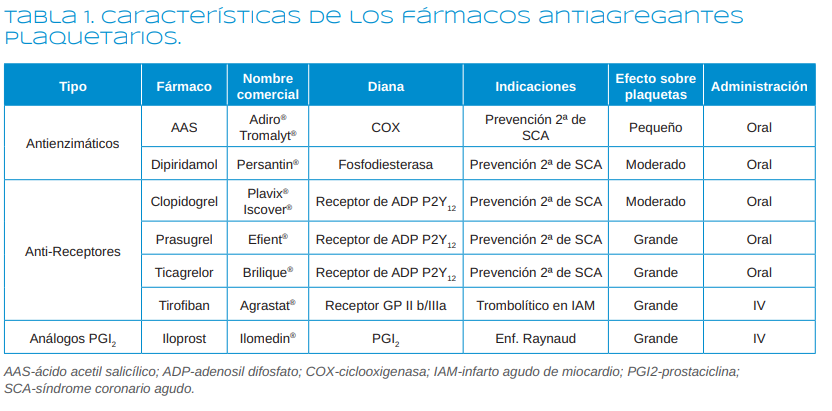
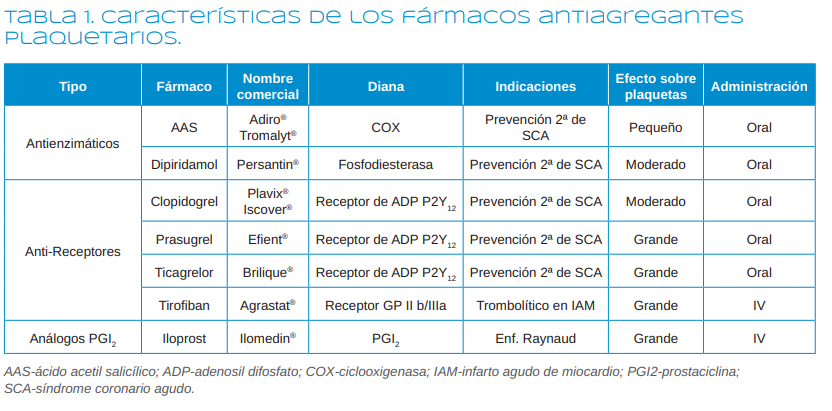
acetilsalicílico) o la fosfodiesterasa (dipiridamol), en bloquear el receptor P2Y12 plaquetario (como las tienopiridinas) o el receptor GP IIb/III a (como el tirofiban) o en actuar como análogos de moléculas que inhiben la agregación plaquetaria (como el iloprost) (Tabla 1).
Los AAP se pueden clasificar según su mecanismo de acción en4-7:
1. Antienzimáticos
1.1. Inhibidores de la ciclooxigenasa (COX)
1.1.1. Ácido acetil salicílico-AAS (Aspirina®, Adiro®, Tromalyt®)
1.1.2. Triflusal (Disgren®)
1.2. Inhibidores de la fosfodiesterasa
1.2.1. Dipiridamol (Persantin®)
1.2.2. Cilostazol
1.2.3. Pentoxifilina
2. Inhibidores de los receptores
2.1. De ADP (P2Y12): Ticlopidina, Clopidogrel, Prasugrel, Ticagrelor
2.2. De GP IIb/IIIa: Abcximab, Tirofiban, Eptifibatide
2.3. De trombina (PAR1): Vorapaxar
3. Análogos de la prostaciclina
3.1. Iloprost (Ilomedin®)
1. Antienzimáticos
1.1. Inhibidores de la COX
1.1.1. Ácido acetil salicílico (Adiro® y Tromalyt®)
El ácido acetil salicílico (AAS) es el antiagregante por excelencia. Es un inhibidor irreversible de las ciclooxigenasas COX-1 y COX-2
de la plaqueta y, por tanto, inhibe la síntesis
del TXA2 plaquetario y de la PGI2 del endotelio vascular, pero sobre todo del primero.
Además, dosis pequeñas parecen afectar solo al TXA2. Asimismo, las plaquetas, al no
tener núcleo, no tienen capacidad de volver a sintetizar la COX, a diferencia de las células endoteliales, por lo que la inhibición del TXA2 dura lo que dura la vida media de la plaqueta, es decir, entre 7 y 10 días.
La inhibición del TXA2 solo suprime uno de los mecanismos de la agregación, pero no afecta a la agregación inducida por ADP.
Sin embargo, otro efecto del AAS en las plaquetas es que disminuye la secreción de los gránulos densos, es decir, disminuye la liberación de sustancias proagregantes durante
la activación plaquetaria.
Esto explicaría por qué sus efectos sobre las plaquetas son mayores de lo que cabría esperar de la simple inhibición plaquetaria dependiente de un agonista relativamente débil como es el TXA21.
El AAS es la terapia antitrombótica básica, usada como tratamiento antiagregante único en la prevención secundaria de la enfermedad aterosclerótica8.
1.1.2. Triflusal (Disgren®)
El triflusal es un análogo del AAS, que inhibe de forma selectiva la COX plaquetaria, pero no afecta a las células endoteliales. El triflusal tiene menos efectos secundarios que el
AAS, por lo que se indica en pacientes con resistencia al AAS y en pacientes geriátricos.
1.2. Inhibidores de las fosfodiesterasas
1.2.1. Dipiridamol (Persantin®)
El dipiridamol es un inhibidor de la fosfodiesterasa, que aumenta los niveles de AMPc intracelular, inhibiendo la agregación; además es vasodilatador. No presenta ventajas respecto al AAS, pero
puede asociarse a fármacos anticoagulantes y
darse a pacientes portadores de prótesis valvulares cardíacas con intolerancia al AAS.
1.2.2. Cilostazol
Incrementa los niveles de AMPc intracelular y es
vasodilatador.
1.2.3. Pentoxifilina
La pentoxifilina es un vasodilatador inhibidor de la
fosfodiesterasa, que actualmente se emplea en la prevención de la osteonecrosis de los maxilares.
2. Inhibidores de los receptores plaquetarios
2.1. Inhibidores del receptor de ADP P2Y12
2.1.1. Inhibidores irreversibles: Tienopiridinas
2.1.1.1. De 1ª generación: Ticlopidina (Tiklid®)
La ticlopidina es un derivado tienopiridínico, que se comporta como un profármaco, es decir, se metaboliza en el hígado dando lugar a un metabolito activo, que antagoniza la agregación inducida por ADP. Fue el primer
inhibidor del receptor P2Y12, pero la relativa frecuencia de reacciones adversas, como la
diarrea y, sobre todo, la neutropenia (en un 0,8% de los casos), ha hecho que su uso sea
cada vez más reducido.
2.1.1.2. De 2ª generación: Clopidogrel (Plavix®,
Iscover®)
Es un profármaco, que requiere dos reacciones de oxidación en el hígado para transformarse en el metabolito activo, que
inhibe al receptor P2Y12. Sin embargo, se ha descrito una gran variabilidad individual en la respuesta antiagregante inducida por clopidogrel. Suele emplearse a una dosis de 75 mg al día, siendo más potente que 100
mg de AAS.
Se puede administrar juntamente con el AAS, para el tratamiento del SCA tras la colocación un stent coronario o posterior a la
cirugía de revascularización percutánea, lo que constituye la llamada “terapia antiagregante dual” o “doble antiagregación plaquetaria” (DAP).
2.1.1.3. De 3ª generación: Prasugrel (Efient®)
El prasugrel es otro profármaco que inhibe el receptor P2Y12, y es más potente, más rápido y presenta menor variabilidad en la respuesta antiagregante que el clopidogrel. Es el único que tiene beneficios en diabéticos.
2.1.2. Inhibidores reversibles
2.1.2.1. Ticagrelor (Brilique®)
El ticagrelor es un antagonista del receptor P2Y12 de efecto reversible. Es más rápido y potente que el clopidogrel. Además, tiene efectos extraplaquetarios que resultan beneficiosos desde un punto de vista cardiovascular.
2.1.2.2. Cangrelor
Recientemente se han diseñado nuevos antagonistas cangrelor y elinogrel, aún más potentes. Estos nuevos antiagregantes consiguen una mayor eficacia antitrombótica, pero también implican un mayor riesgo de
sangrado.
2.2. Inhibidor del receptor GP IIb/IIIa
2.2.1. Abcximab, Tirofiban, Eptifibatide
Son antiagregantes de uso hospitalario que se
emplean de forma IV, bloqueando la unión del fibrinógeno y el FVW a las glicoproteínas de la
superficie plaquetaria (mediada por el receptor GP II b/III a).
Se emplean como fármacos trombolíticos de urgencia en el tratamiento del IAM. Cuanto antes se instaure la terapia con inhibidores del GP IIb/III a, más favorable será el pronóstico del IAM.
2.3. Antagonista del PAR1 (receptor activador de proteasa 12)
2.3.1. Vorapaxar (Zontivity ®)
Inhibe la agregación mediada por la trombina, pues es antagonista del receptor de trombina PAR1. Aceptado por la FDA, pero no por la EMA.
3. Análogos de la prostaciclina
3.1. Iloprost (Ilomedin®)
El iloprost es un análogo de la prostaciclina, que aumenta el AMPc intraplaquetario y además es vasodilatador. Se usa en arteriopatías periféricas, tromboangeítis obliterante y enfermedad de Raynaud.