Caso clínico
Pénfigo Vulgar mucoso: a propósito de un caso. Revisión de la literatura
Introducción: El Pénfigo Vulgar (PV) es una enfermedad de origen autoinmune caracterizada por causar ampollas intraepidérmicas en piel y mucosas, como consecuencia de la agresión de autoanticuerpos hacia varios tipos de proteínas desmosómicas. El elemento eruptivo primordial es la ampolla, que puede presentarse de manera individual o en coalescencia con la consecuente formación de placas erosivo-costrosas. En el 90% de los casos las lesiones afectan a la mucosa oral, mientras que en el 50-70% de los mismos constituyen la primera manifestación de la enfermedad.
Objetivo: Se presenta un caso clínico de PV y una revisión bibliográfica actualizada, con el objetivo de analizar sus factores etiológicos y sus opciones terapéuticas.
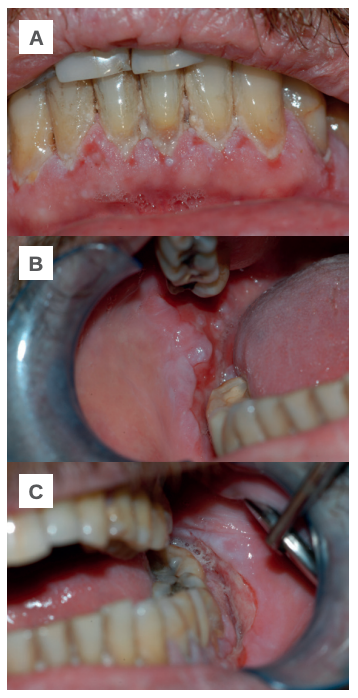
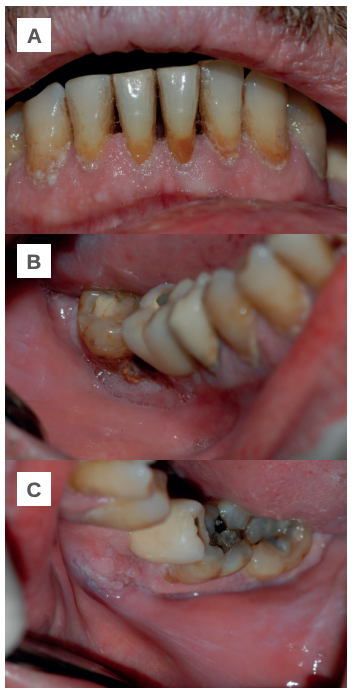
Caso clínico: Se presenta el caso de un paciente varón, de 71 años, fumador, con condición prediabética y síndrome de Guillain Barré remitido al Servicio de Cirugía Bucal e Implantología del Hospital Virgen de la Paloma de Madrid con un cuadro clínico caracterizado por infección oral y pérdida de peso. Una vez confirmado el diagnóstico de PV mucoso mediante examen histopatológico, se estableció una terapia con corticoides sistémicos obteniendo una remisión casi completa de las lesiones. Tras varias semanas de tratamiento su dermatólogo decidió suspender los corticoides para llevar a cabo, sin éxito, una terapia sustitutiva con inmunosupresores. La recidiva de las lesiones, unida a los efectos adversos causados por la nueva terapia, obligó a reconsiderar la suministración de corticoides con una resolución positiva de la enfermedad.
Conclusiones: No existe una estrategia terapéutica bien establecida para el tratamiento del PV. El objetivo del tratamiento consiste en conseguir una remisión clínica prolongada mediante la suministración de corticoesteroides. El tratamiento con inmunosupresores (azatioprina, micofenolato mofetil) no ha demostrado ser una alternativa válida a los corticoides, pero sí puede ser una buena opción como tratamiento adyuvante con el fin de reducir las dosis de corticoides.
Introduction: Pemphigus Vulgaris is an autoimmune disease characterized by causing intraepidermal blisters on the skin and mucosa, as a consequence of the aggression of autoantibodies towards various types of desmosomal proteins. The primary eruptive element is the blister, which can appear in coalescence with the consequent formation of erosive-crusted plaques. In 90% of cases lesions affect the oral mucosa, while in 50-70% they are the first manifestation of the disease.
Objective: We aim to report a case of Pemphigus Vulgaris and an updated literature review to analyse its etiological factors and treatment options.
Clinical case: We present the case of a 71-year-old male patient, smoker, with prediabetic condition and sindrome Guillain Barré referred to the Oral Surgery and Implantology Service of the Virgen de la Paloma Hospital in Madrid with a clinical picture characterized by oral infection. Once the diagnosis of mucosal PV was confirmed, a systemic corticosteroid therapy was established, obtaining almost complete remission of the lesions. After several weeks of treatment, his dermatologist decided to suspend the corticosteroids to carry out unsuccessful immunosuppressant replacement therapy. The recurrence of the lesions, together with the adverse effects caused by the new therapy, forced the reconsideration of the supply of corticosteroids with a positive resolution of the disease.
Conclusions: There is no definitive therapeutic strategy for the treatment of Pemphigus Vulgaris. The goal of treatment is to achieve a prolonged clinical remission by supplying corticosteroids. Immunosuppressants have not been shown to be a valid alternative to corticosteroids, but they are a good option if they are supplied together with corticoids in order to reduce their doses.
El término pénfigo reúne un grupo de enfermedades autoinmunes caracterizadas por causar ampollas intraepidérmicas en piel y mucosas, como consecuencia de la agresión de autoanticuerpos hacia varios tipos de proteínas desmosómicas. En base a las manifestaciones histopatológicas, así como a los tipos de autoanticuerpos y antígenos implicados, se distinguen varias formas de pénfigo: vulgar, cicatricial, foliáceo, paraneoplásico, herpetiforme e IgA.
Clínicamente el elemento eruptivo primordial es la ampolla, que puede presentarse de manera individual o en coalescencia con la consecuente formación de placas erosivo-costrosas. En el 90% de los casos las lesiones afectan a la mucosa oral, mientras que en el 50-70% de los mismos constituyen la primera manifestación de la enfermedad1 . Es una patología de curso progresivo que, en sus formas más graves y sin un adecuado tratamiento, puede provocar un fuerte desequilibrio electrolítico, sepsis e insuficiencia cardiaca2 .
Desde una perspectiva epidemiológica, el pénfigo muestra una distribución desigual según factores étnicos, geográficos y sexuales. Así, mientras que en Europa Central la incidencia de Pénfigo Vulgar (PV) es de 0,5 nuevos casos por millón de habitantes al año, en Israel las cifras llegan hasta los 16,1 casos por millón al año, afectando en particular modo a los judíos de la etnia Ashkenazi3-4. Varios estudios señalan una mayor propensión por el género femenino, con una proporción mujer:hombre que oscila entre 1:1 en Finlandia y 5:1 en EEUU5 .
Si bien es cierto que las tasas de mortalidad actuales raramente superan el 5% en los países desarrollados, es importante recordar que se trata de una afección potencialmente mortal y cuyos signos se pueden detectar de forma temprana en la consulta odontológica6 . El dolor, junto con la necesidad de emprender tratamientos de larga duración, obligan a estos pacientes a acudir con frecuencia a especialistas capaces de mejorar su calidad de vida.
El objetivo de esta publicación es la descripción de un caso de PV mucoso limitado a la región oral, exponiendo las últimas actualizaciones sobre la etiología, las manifestaciones clínicas, el diagnóstico y el tratamiento de la enfermedad.