Puesta al día
Consideraciones bucodentales en pacientes con Síndrome de Morquio
La mucopolisacaridosis tipo IV (MPS-IV) también conocida como enfermedad de Morquio en recuerdo del pediatra uruguayo Luis Morquio que la describió por primera vez, es una enfermedad congénita causada por la deficiencia de la enzima N-acetilgalactosamina 6 sulfatasa o de la enzima B-Galactosidasa. Estas anomalías enzimáticas tienen como consecuencia que se acumulen en diferentes tejidos del organismo cantidades elevadas de mucopolisacaridos.
En la bibliografía se describe con detalle los defectos del esmalte que presentan los pacientes diagnosticados del síndrome de Morquio. Estos defectos son una característica aparentemente constante en la enfermedad y, por lo tanto, hace necesaria las visitas al odontólogo para su control evitándose problemas mayores. Dichos defectos consisten en un esmalte anormalmente delgado, que es áspero debido a los numerosos hoyos diminutos y a una superficie irregular. La delgadez del esmalte da como resultado una forma alterada y decoloración de los dientes que, añadido a los diastemas interdentales, provocan alteraciones en la oclusión. Aparte de estos defectos, el esmalte es histológicamente normal y tiene una dureza y radiodensidad normales. El tratamiento odontológico de los pacientes con MPS-IV requiere colaboración multidisciplinar, debido a que las manifestaciones orales de la enfermedad pueden aparecer a cualquier edad, resultando en ocasiones tedioso para el paciente y complicado para el profesional. Especial mención merecen las terapias utilizadas como tratamiento sintomático de la enfermedad, así como el manejo de la vía aérea en el caso de intervenciones bajo anestesia general o sedación para tratar ciertas patologías del territorio bucomaxilodental.
Mucopolysaccharidosis type IV (MPS-IV) also known as Morquio’s disease in memory of the Uruguayan pediatrician Luis Morquio who described it for the first time, is a congenital disease caused by the deficiency of the enzyme N-acetylgalactosamine 6 sulfatase or enzyme B -Galactosidase. These enzymatic anomalies result in high amounts of mucopolysaccharides accumulating in different tissues of the organism.
The enamel defects presented by patients diagnosed with Morquio syndrome are described in detail in the bibliography. These defects are an apparently constant feature in the disease and, therefore, make visits to the dentist necessary for their control, avoiding major problems. These defects consist of an abnormally thin enamel that is rough due to numerous tiny holes and an irregular surface. The thinness of the enamel results in an altered form and discoloration of the teeth, which added to the interdental diastemas, cause alterations in the occlusion. Apart from these defects, the enamel is histologically normal and has a normal hardness and radiodensity.
Dental treatment of patients with MPS-IV requires multidisciplinary collaboration, because the oral manifestations of the disease can appear at any age, being sometimes tedious for the patient and complicated for the professional. Special mention should be made of the therapies used as a symptomatic treatment of the disease, as well as the management of the airway in the case of interventions under general anesthesia or sedation to treat certain pathologies of the bucomaxillodental territory.
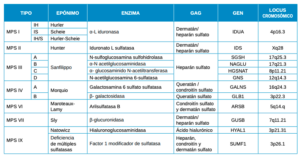
Las mucopolisacaridosis (MPS) son un grupo de trastornos de almacenamiento lisosomal caracterizados por la deposición tisular de glicosaminoglicanos (GAG), anteriormente denominados mucopolisacáridos. Esto es debido, a un defecto enzimático en las vías de degradación de los GAG, que en función de la enzima ausente o cuya actividad esté mermada, dará lugar a los diferentes tipos de mucopolisacaridosis (Tabla 1).
Como en la mayoría de las MPS, no se observa sintomatología en el neonato y, se comienza a sospechar de estos trastornos cuando los niños tienen entre 1 y 3 años, no siendo hasta los 5 años de media cuando se tiene la confirmación del diagnóstico. Los primeros casos de MPS IV fueron descritos por el pediatra uruguayo Luis Morquio, cuando en el año 1929 comunicó el caso de cuatro hermanos de una misma familia con distrofia esquelética severa1 . En ese mismo año James Frederick Brailsford, radiólogo inglés, describió un paciente de cuatro años de edad con la misma condición, dándole el nombre de osteocondrodistrofia. Morquio especificó cambios radiológicos observables antes de la aparición de los signos y síntomas clínicos que suelen debutar cuando el paciente tiene tres años2 .
Se trata, pues, de una enfermedad rara autosómica recesiva, con una incidencia variable según el origen geográfico y étnico que oscila entre 1/76.000 de los recién nacidos vivos en Irlanda del Norte hasta 1/450.000 de los nacidos vivos de Holanda y Portugal en el subtipo A, e incluso 1/640.000 en su variante más rara (subtipo B)3-5.
La esperanza de vida depende de la gravedad de la enfermedad, presentando un margen amplio que oscila entre los 20 y los 70 años. La muerte, generalmente, acontece debido a insuficiencia cardiorrespiratoria por deformidad del esqueleto torácico que compromete la vía aérea, o bien por compresión del canal espinal por luxación a nivel de la apófisis odontoides.
En las enfermedades de almacenamiento lisosómico, alguna enzima del lisosoma tiene actividad reducida o nula debido a un error genético, por lo que el sustrato de dicha enzima comienza a acumularse de manera progresiva y se deposita dentro de los lisosomas, que aumentan de tamaño interfiriendo con los procesos celulares6, 7.
Actualmente existe una distinción entre dos variantes de la misma enfermedad (Figura 1).
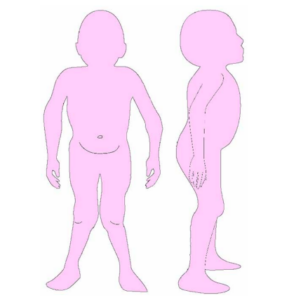
El tipo “A” es el fenotipo más grave que se manifiesta, principalmente, como una displasia esquelética progresiva debido a la alteración del metabolismo del queratán y condroitín sulfato por ausencia de la enzima galactosamina-6-sulfatasa afectando a nivel de fibroblastos y leucocitos.
El 25% de los afectados por este síndrome presentan la variante más atenuada (MPS IV tipo “B”) que, si bien tienen mutaciones definidas y demostrables en el mismo gen, presentan rasgos mucho más leves y menos marcados por alteración del metabolismo del queratán sulfato presentando menor afectación corneal y cardiaca debido a déficit de la enzima beta-galactosidasa8 .
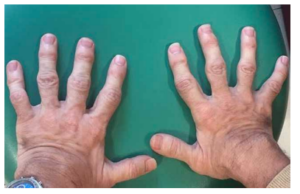
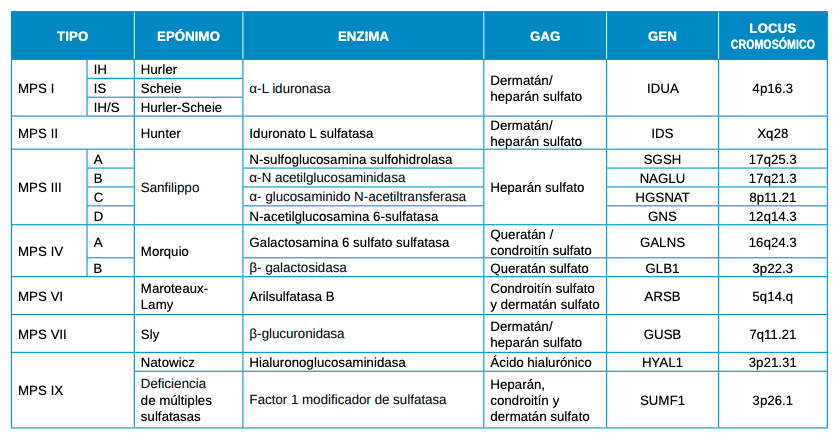
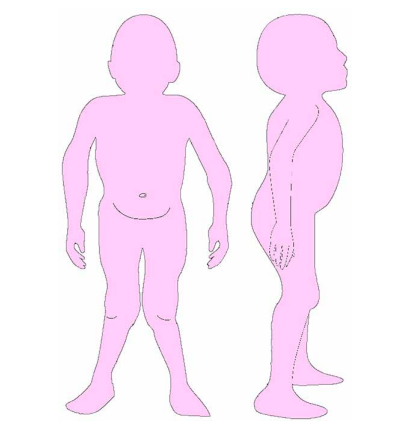
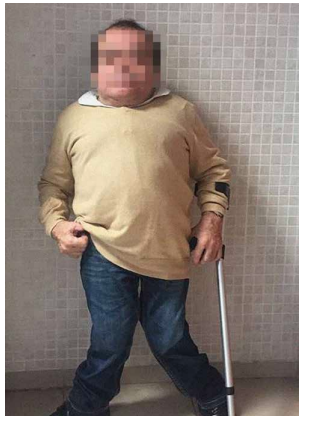
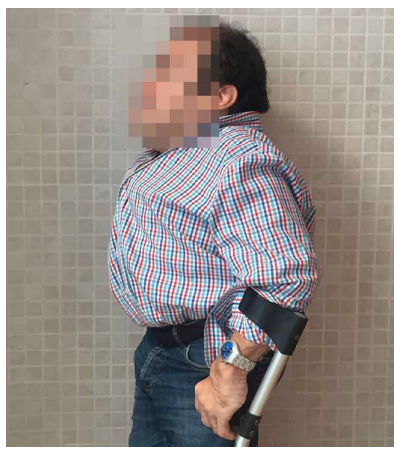
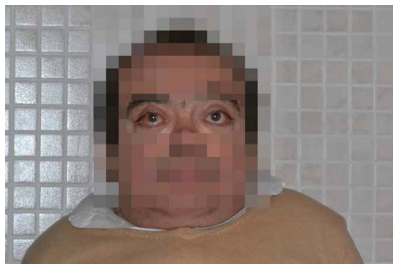
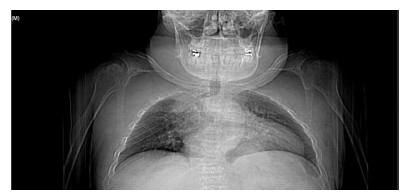
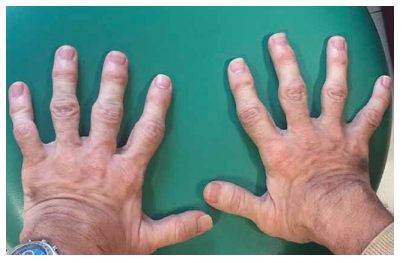
Las características físicas pueden conducir a identificar un fenotipo reconocible con baja estatura desproporcionada, con tronco marcadamente corto, cuello corto, pectus carinatum (con forma de barril), brazos y piernas delgados, contracturas en la flexión de las caderas, pies valgos y engrosamiento facial (Figuras 2-6).
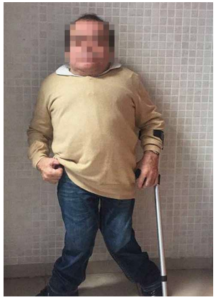
miembros inferiores.
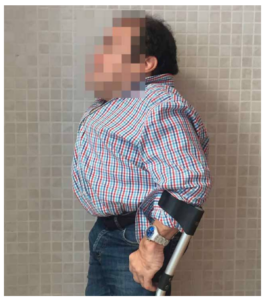

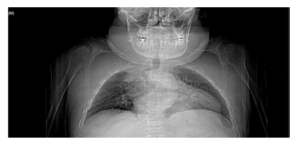
Se pueden identificar manifestaciones clínicas esqueléticas en donde al interrumpirse el desarrollo normal y la maduración del cartílago y el hueso aparecen malformaciones y laxitud articular con inestabilidad cervical9, 10-19.
Es evidente la aparición de artritis prematura, hipermovilidad articular sobre todo en muñecas y tobillos, cifosis y cifoescoliosis (Figura 7).
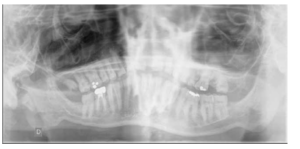
Generalmente el tamaño del paciente al momento del nacimiento es normal, deteniéndose el crecimiento a muy temprana edad (7-12 años), con lo que la estatura promedio en la edad adulta oscila entre 122 cm en varones y 113 cm en mujeres.
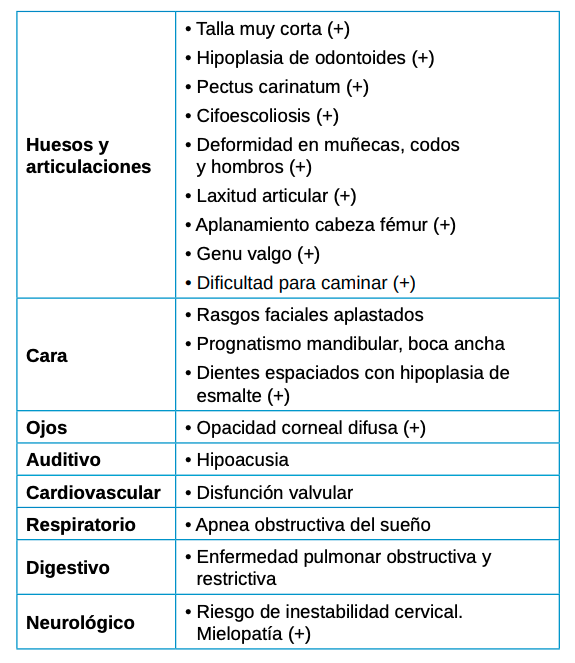
Se deben tener en cuenta las manifestaciones extraesqueléticas por su importante contribución a la progresión de la enfermedad y, por consiguiente, su impacto en la calidad de vida del paciente (Tabla 2).
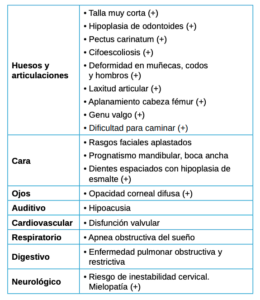
Dentro de éstas, las más importantes mencionadas en la literatura son las siguientes20, 21-24:
- A nivel ocular, la opacidad corneal es menos frecuente y más lentamente progresiva lo que implica una disminución de la agudeza visual la existencia de fotofobia
- A nivel auditivo, con pérdida de audición mixta, tanto conductiva como neurosensorial, de manera moderada y en casos más severos sería necesario el uso de audífonos.
- A nivel cardiovascular, muy común la enfermedad coronaria leve que rara vez desarrollan cardiopatía. Pero si existe incompetencia valvular deberán tomarse las medidas preventivas oportunas.
- A nivel respiratorio, la enfermedad pulmonar restrictiva puede ser secundaria al menor tamaño de la caja torácica, y en casos de obstrucción valorar tonsilectomía o adenoidectomía. Importante mencionar también las infecciones respiratorias recurrentes, así como la apnea obstructiva del sueño y la traqueomalacia.
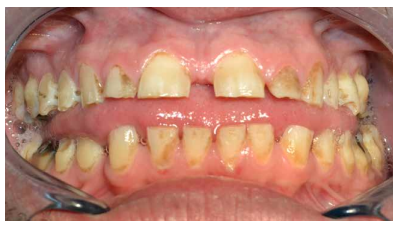
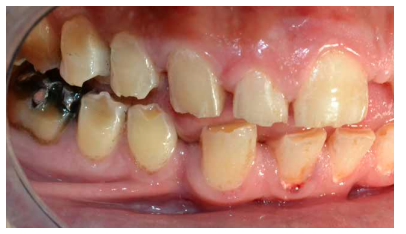
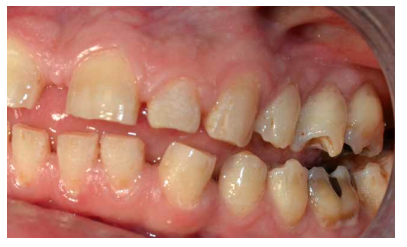
- A nivel dentario, el esmalte es uniformemente anormal, delgado, rugoso e hipoplásico. Afecta a ambas denticiones, y provocando una marcada frecuencia de fracturas dentarias, desgastes o abrasiones y caries. Existe un importante componente lingual en maloclusión, diastemas en toda la arcada y prognatismo mandibular, así como bruxismo y patología de ATM. Incisivos con forma de pala y molares cóncavos que favorece las lesiones de caries y sensibilidad dentinaria.
- Problemas de obesidad, debido a un bajo nivel de actividad que en muchos casos reduce la movilidad al máximo siendo imprescindible el uso de silla de ruedas desde la adolescencia.
Las anomalías musculoesqueléticas y el almacenamiento de GAG en las vías respiratorias aumentan el riesgo de los pacientes sometidos a anestesia. Estudios como el de Theroux y cols.,25 Walker y cols.,26 y Chaudhuri y cols.,27 evalúan un enfoque perioperatorio y postoperatorio tras la administración de sedantes o anestéticos intravenosos, en relación al mantenimiento de las vías respiratorias e informa de los hallazgos positivos obtenidos mediante broncoscopia flexible, en la vía aérea superior e inferior durante estos procedimientos. Son, por tanto, enfermedades multisistémicas que requieren un enfoque multidisciplinar para su tratamiento y manejo adecuado.
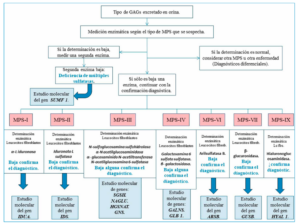
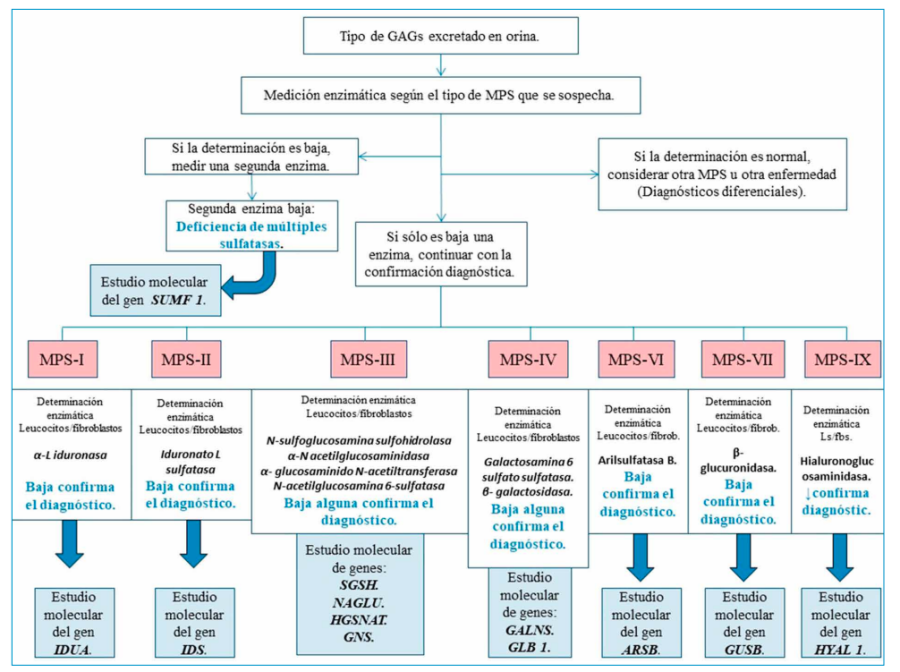
Ante la sospecha clínica de encontrarse ante una MPS, la primera prueba a realizar es una determinación de GAG en orina. Si se hallan en cantidades normales se podrá descartar casi con total seguridad que se trate de este tipo de enfermedad.
Si por el contario, hay una determinación positiva en orina de glicosaminoglicanos, el siguiente paso será determinar la actividad enzimática de aquella que sospechemos como causa de la enfermedad28.
Esta determinación se suele realizar en leucocitos o fibroblastos aunque la técnica más óptima y adecuada es la prueba de manchas de sangre seca (Dryed Blood SpotsDBS), consistente en espectrometría de tándem de masas en gota de sangre seca. Una actividad enzimática menor de 10% es muy sugestiva de MPS, y si fuese menor del 2% es diagnóstico de la misma20-28.
Por último, se debería realizar diagnóstico genético y localizar la mutación génica responsable de la enfermedad. A todo este proceso es a lo que se conoce como algoritmo diagnóstico en MPS (Figura 8).
En cuanto al tratamiento, hasta el momento actual ha sido encaminado fundamentalmente en cuidados de soporte tales como AINEs para el dolor y artritis; bifosfonatos para osteoporosis; oxigenoterapia y la presión positiva continua en la vía aérea (Continuous Positive Airway Pressure-CEPAP); numerosas intervenciones quirúrgicas; fisioterapia en todas sus versiones; tubos de drenaje tubárico; adenoidectomía; amigdalectomía; herniorrafias y patología valvular28, 29.
Hay escasa experiencia en el trasplante de progenitores hematopoyéticos, pero los estudios existentes no dan evidencia satisfactoria de su beneficio. La terapia génica aún se encuentra en etapas preclínicas, y existe el fármaco llamado Vimizim® (terapia de reemplazo enzimático), producido por recombinación en una línea de células humanas con objeto de reducir el acúmulo de queratán sulfato y, así mejorar los síntomas y signos de la enfermedad, penetrando en la placa del cartílago de crecimiento26-30.
Se debe realizar un seguimiento y monitorización periódica de estos pacientes para valorar la evolución y poder controlar, y prevenir la aparición de complicaciones y evaluar la efectividad de los tratamientos aplicados. Esto incluye también el ámbito bucodental, las revisiones periódicas y actuaciones preventivas tanto a nivel cariológico como a nivel periodontal31, 32.
Según la experiencia de algunos autores32-38, corresponden hasta en un 69% de las alteraciones extraesqueléticas y se debe dar un enfoque multidisciplinar y cirugías combinadas a la vez que se brinda un manejo seguro de la vía aérea, incluso para evaluaciones de diagnóstico de modo que se pueda conseguir una mayor atención al paciente sin riesgos adicionales33, 34.
Es posible que se produzca un compromiso significativo de las vías aéreas por multitud de factores en la primera infancia, incluidos los depósitos de GAG de las vías aéreas superiores e inferiores35.
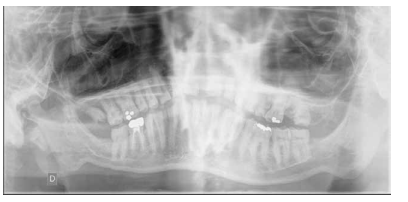
En la mayoría de los niños se pueden observar características dentales como cúspides puntiagudas, incisivos en forma de pala, esmalte delgado y superficies bucales con hoyos. Además, la ATM puede verse afectada con una reabsorción severa de la cabeza del cóndilo36 (Figuras 9-11).
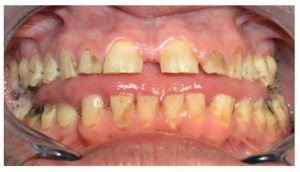
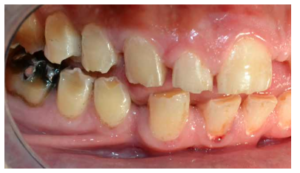
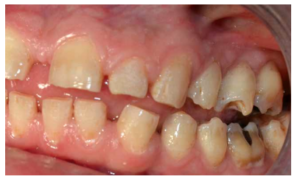
dentales.
El examen histológico de los molares primarios exfoliados mostrará una banda de porosidad aumentada a continuación de las estrías de Retzius en la parte exterior del esmalte. Estas alteraciones del desarrollo se asocian ocasionalmente con defectos menores localizados en la superficie del esmalte37.
Conviene enfatizar además la importancia de una vigilancia cercana al desarrollo dental y al cuidado bucal de manera regular para prevenir el desgaste de los dientes, la pérdida de la altura vertical de la superficie oclusal y el riesgo subsecuente de disfunción de la articulación temporomandibular (ATM).
Los pacientes con síndrome de Morquio, a menudo, necesitan tratamiento de ortodoncia para la corrección de la maloclusión, lo que a su vez mejora la función masticatoria. Se podrían corregir las inclinaciones labiales de los dientes anteriores superiores e inferiores y se cerrarían los espacios interdentales para una correcta higiene, evitándose así la aparición de caries37.
Se han formulado hipótesis sobre la posibilidad de que el tratamiento de ortodoncia temprano y la observación a largo plazo en un paciente con síndrome de Morquio pueden mejorar de manera considerable el curso de los trastornos musculoesqueléticos de la ATM. Hasta la fecha, no existen informes de casos que describan el tratamiento precoz de ortodoncia en dichos pacientes, pero cabe esperar que como en cualquier paciente en edad infantil, dichos tratamientos además de ser menos agresivos, sean más eficaces y rápidos que en el adulto38.
Estos pacientes muestran arcos maxilares y mandibulares espaciados con un hábito de empuje lingual a la vez que también suelen presentar un labio superior sobresaliente, inclinación labial de los dientes anteriores superiores e inferiores y esmalte delgado. El hábito de empuje de la lengua suele desaparecer después de la aplicación de un aparato de ortodoncia removible. La dentición espaciada en los arcos superior e inferior mejora después del tratamiento con un dispositivo fijo con cable de arco de bucle de cierre (0.017 x 0.025) y bandas en los molares superiores e inferiores, sin utilizar dispositivos de borde debido a la fragilidad y mala calidad del esmalte38.
El labio superior sobresaliente y la inclinación labial de los dientes anteriores superiores e inferiores también mejoraran después del tratamiento, pero no se logra la intercuspidación óptima de los dientes. Estudios como el de Meza Cabrera y cols.,39 o bien como el de Kuratani y cols.,40 sugieren que el tratamiento de ortodoncia temprana podría mejorar la maloclusión en un paciente con síndrome de Morquio, pudiendo lograr una mejoría de la función masticatoria durante un período de retención a largo plazo.
Por otro lado, cabe mencionar que estos pacientes debido a los problemas musculoesqueléticos habitualmente están en tratamiento con bifosfonatos, por lo que conviene extremar las precauciones a la hora de realizar tratamientos quirúrgicos los cuales deben realizarse de la manera más atraumática posible, y bajo profilaxis antibiótica.
Además, en los casos de afectación valvular severa conviene realizar profilaxis de la endocarditis previa a tratamientos dentales invasivos mediante la pauta habitual de 2 gr de amoxicilina 1 hora antes de la intervención, y en alérgicos a la penicilina, 600 mg de clindamicina41.
Debido a la talla baja y alteraciones en columna cervical quizás sea necesario adaptar al sillón dental accesorios o cojines para mayor comodidad del paciente en tratamientos prolongados asegurándonos de mantener permeable la vía aérea y sin hiperextender demasiado la columna cervical.
Hay que tener en cuenta en la administración de fármacos, ya sea tanto por vía oral para controlar el postoperatorio, como en casos de sedación o anestesia general, que las dosis deberán ajustarse al peso e índice de masa corporal (IMC) del paciente.
En conclusión, hasta el día de hoy, no existe ninguna cura para individuos afectados de estos desórdenes, pero existen maneras de manejar los diferentes retos y problemas de salud que tendrán tanto pacientes como familiares. El trasplante de médula ósea ha sido utilizado para el tratamiento de MPS IV, pero con muy poco éxito. La comunidad científica continúa buscando una forma mejor y más efectiva para tratar estos desórdenes para darles más oportunidades a estos individuos en el futuro.

Ruíz Sáenz, Pedro Luis
Unidad de Estomatología, Hospital Central de la Cruz Roja, Madrid.
López Rodríguez, Mónica A.
Unidad de Enfermedades Sistémicas y Minoritarias, Servicio de Medicina Interna, Hospital Ramón y Cajal, Madrid.
Sanz Alonso, Javier
Profesor Asociado de Cirugía Bucal Universidad Complutense de Madrid (UCM).
Buesa Barez, José María
Profesor Asociado del Máster de Cirugía Bucal e Implantología, Hospital Virgen de la Paloma.
Martínez González, José María
Profesor Titular de Cirugía Bucal y Maxilofacial UCM.
Indexada en / Indexed in: – IME – IBECS – LATINDEX – GOOGLE ACADÉMICO